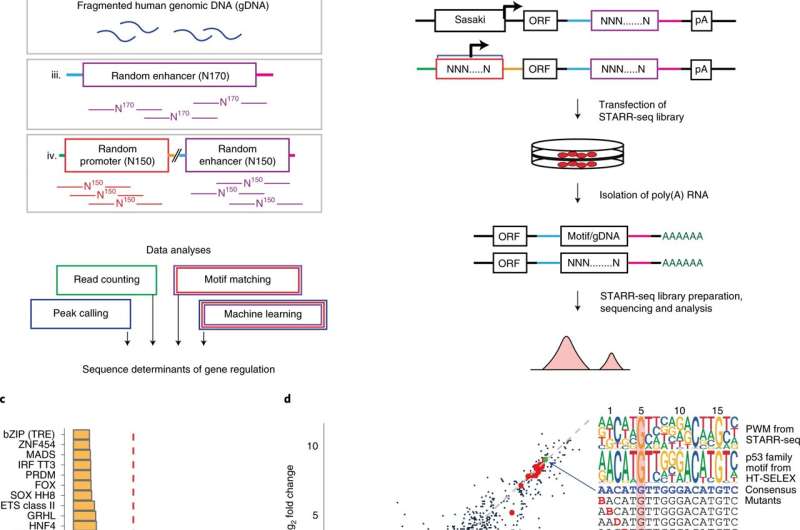
 Methods, Supplementary Note and Supplementary Tables 3 and 4). b, MPRA (STARR-seq) reporter construct and its variations, and the experimental workflow for measuring promoter or enhancer activity. The MPRA libraries are transfected into human cells, and RNA is isolated 24 h later, followed by enrichment of reporter-specific RNA, library preparation, sequencing and data analysis. The active promoters are recovered by mapping their transcribed enhancers to the input DNA and identifying the corresponding promoter. c, Enhancer activity of HT-SELEX motifs measured from the synthetic TF motif library in GP5d cells. Median fold change of the sequence patterns containing a single instance of the motif consensus or its reverse complement over the input library is shown. Red line marks 1% activity related to the strongest motif. Dimeric motifs are indicated by orientation with respect to core consensus sequence (GGAA for ETS, ACAA for SOX, AACCGG for GRHL and GAAA for IRF; HH, head to head; HT, head to tail; TT, tail to tail), followed by gap length between the core sequences. Asterisk indicates an A-rich sequence 5′ of the IRF HT2 dimer. Supplementary Table 5 describes the naming of the motifs in each figure. d, The effect of a mismatch on enhancer activity of the p53 family (p63) motif when a consensus base is substituted by any other base one position at a time. The log2 fold change compared to input is plotted for the same motif pattern in two different sequence contexts. The PWMs for HT-SELEX and STARR-seq motifs are shown; note that mutating G to any other base (H) at position 5 (H05) leads to almost complete loss of activity. Credit: DOI: 10.1038/s41588-021-01009-4" width="800" height="530">
Methods, Supplementary Note and Supplementary Tables 3 and 4). b, MPRA (STARR-seq) reporter construct and its variations, and the experimental workflow for measuring promoter or enhancer activity. The MPRA libraries are transfected into human cells, and RNA is isolated 24 h later, followed by enrichment of reporter-specific RNA, library preparation, sequencing and data analysis. The active promoters are recovered by mapping their transcribed enhancers to the input DNA and identifying the corresponding promoter. c, Enhancer activity of HT-SELEX motifs measured from the synthetic TF motif library in GP5d cells. Median fold change of the sequence patterns containing a single instance of the motif consensus or its reverse complement over the input library is shown. Red line marks 1% activity related to the strongest motif. Dimeric motifs are indicated by orientation with respect to core consensus sequence (GGAA for ETS, ACAA for SOX, AACCGG for GRHL and GAAA for IRF; HH, head to head; HT, head to tail; TT, tail to tail), followed by gap length between the core sequences. Asterisk indicates an A-rich sequence 5′ of the IRF HT2 dimer. Supplementary Table 5 describes the naming of the motifs in each figure. d, The effect of a mismatch on enhancer activity of the p53 family (p63) motif when a consensus base is substituted by any other base one position at a time. The log2 fold change compared to input is plotted for the same motif pattern in two different sequence contexts. The PWMs for HT-SELEX and STARR-seq motifs are shown; note that mutating G to any other base (H) at position 5 (H05) leads to almost complete loss of activity. Credit: DOI: 10.1038/s41588-021-01009-4" width="800" height="530"> The logic that controls gene regulation in human cells has been discovered by a research group. In the future, this knowledge could be used to investigate cancer and other genetic diseases.
Gene regulation controls the activity of genes in cells. Incorrect gene regulation can lead to diseases.
The human genome contains genes that are used to make muscles and brain cells that can process information. The genes in the brain and muscles are regulated by the genes in the DNA.
The regulatory code that determines gene activity is poorly understood. The human genome contains almost 3 billion base pairs, but it is too short for learning the regulatory code. The problem is similar to that faced by a linguist who tries to understand a forgotten language on the basis of a few short texts.
A research group of professor Jussi Taipale has found a way around the regulatory code.
The study was published in the journal Nature Genetics.
The first author of the study says that they measured the gene regulatory activity from a collection of DNA sequences that together are 100 times larger than the human genome.
Instead of using the natural genomic sequence, we introduced random synthetic DNA to human cells. Sahu explains that the cells themselves were allowed to read the new DNA and highlight the sequences that function as active regulatory elements.
The key atomic unit of gene expression is identified by researchers.
The researchers used a technique known as massively parallel reporter assays, where the regulatory activity of millions of DNA sequences can be simultaneously studied in one large-scale assays. Artificial intelligence tools were used to analyze the data.
Gene expression is regulated by a number of factors. The key atomic unit of gene expression can be found in the very short DNA sequence to which these factors bind. Individual transcription factors contribute to the regulation of genes. Each factor increases regulatory activity on its own. There are several parallel functions of transcription factors in the gene regulatory process, such as enhancing the rate of gene expression or defining the genomic location where the transcription starts.
Professor Jussi Taipale says that the binding motifs of transcription factors can be thought of as words that together define the cellular gene regulatory code.
The researchers found that most words can be placed in almost any order without changing their meaning.
In some cases similar to compound words, the grammar is strong, and specific combinations of factors need to bind in a certain order.
There are a few highly active transcription factors in cells.
The researchers compared three different human cell types, including colon and liver cancer cells and normal cells from the retina. Only a few transcription factors are active in cells. Regardless of cell type, most transcription factor activities are the same.
The results show that the human cells can be classified into different types based on the context in which they are located.
Traditionally, regulatory elements have been thought to be located in open chromatin regions, where it is easy to access transcription factors. One of the new observations of the study is the discovery of active regulatory elements that function in closed regions. The researchers identified regulatory elements that are dependent on chromatin. These elements are active at their normal sites in the genome, but if they are removed from their original location and transferred to another, their activity will plummet.
More information: Biswajyoti Sahu et al, Sequence determinants of human gene regulatory elements, Nature Genetics (2022). DOI: 10.1038/s41588-021-01009-4 Journal information: Nature Genetics Citation: A study uncovers the 'grammar' behind human gene regulation (2022, February 21) retrieved 21 February 2022 from https://phys.org/news/2022-02-uncovers-grammar-human-gene.html This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no part may be reproduced without the written permission. The content is provided for information purposes only.All Rights Reserved © 2024