The mouse brain has a single-cell definition of genetic identity.
Rachel C. Bandler had a number.
Ilaria Vitali orcid.org/0000-0001-9536-02921 na1.
Ryan N. Delgado is a member of the orcid.org.
May C. Ho orcid.org.
Elena Dvoretskova is a person.
Ibarra Molinas is the son of Josue S. Ibarra.
Paul W. Frazel2.
Maesoumeh Mohammadkhani2.
Robert Machold2.
Sophia Maedler7.
There is a website called orcid.org that is headed by a person named Shane A. Liddelow.
Tomasz J. Nowakowski orcid.org/0000-0003-2345-49644
...
Gord Fishell orcid.org/0000-0000-9640-92783,10
The orcid.org has a biography of Christian Mayer.
This article wasCited by Nature.
The final rounds of cell division give rise to a wide spectrum of postmitotic neurons and glia in the mouse brain. There is an open question about the link between developmental and cell-type diversity. We used parallel tagging to track progenitors during mouse forebrain development. We found diverse types of GABAergic neuron that share a common lineage after we quantified clonal divergence and convergence across all major cell classes. During embryogenesis, the divergence of GABAergic clones occurred, suggesting that the process of differentiation into subtypes is dependent on the progenitor cell level.
The central nervous system has a wide range of different types of neurons and glia. The diversity of the genes in the progenitors is reflected in their regional expression. After cell-cycle exit, transcriptional signatures that distinguish mature neuronal subtypes emerge. The extent to which the trajectory of development is dictated by specific progenitors during the early stages of development remains an open question.
Although previous analyses have shown the distribution of clones, they didn't give any information about their sister cells. The recording of genes and their annotations in in-vivo systems and in mouse embryo has been made possible by the recent discoveries of cellular barcoding strategies and single-cell sequencing.
Here we combined high-throughput single-cellRNA-seq with parallel tagging of progenitors to reconstruct their relationships during the forebrain. We focused our analysis on the gyabaergic neurons, which showed a surprising degree of divergence among different types of inhibitory neurons. After the cell-cycle exit, the giabby neurons that originated from the same progenitor deviated into different paths. Our findings show that the process of differentiating into gyberergic subtypes is dependent on the progenitor cell level.
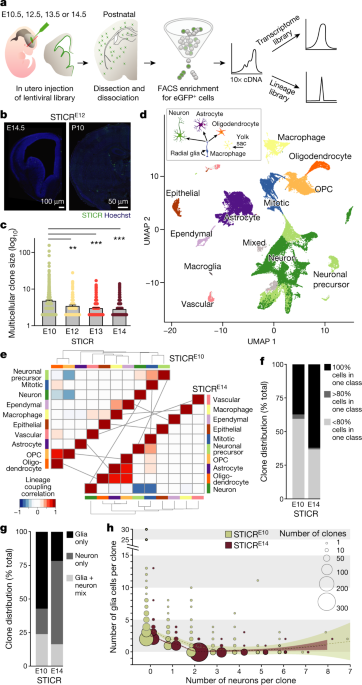
The mouse forebrain has diverse cell types, and we first implemented a lentiviral barcoding method called STICR. The peak of the peak stages of the library were at the beginning of the embryo at the beginning of the embryo at the beginning of the embryo at the beginning of the embryo at the beginning of the embryo at the beginning of the embryo at the beginning of the embryo at the beginning of the embryo The cortex, hippocampus, and olfactory bulb are some of the cells that migrated throughout the forebrain. We waited until the cells were differentiated into mature cell types, then we used the 10x Chromium System to perform scRNA-seq, and then we took the cells to the forebrain tissue to get GFP expression. We analysed transcriptomes from 65,700 cells. To group cells on the basis of patterns of gene expression, we performed a principal components analysis, normalized the replicates, and tracked their position. When the lentiviral library was introduced at E10.5, the average and maximum size of multicellular clones was larger.
There are forebrain cell classes determined via simultaneous capture of transcriptome and barcodes from single cells.
A schematic and images of brain sections injected with STICR are included. The bars indicate time. Each dot is a clone. P 2 and Neurod6 were removed from the one-way analysis of variance. Seurat standard procedures were used to remove clusters expressing marker genes for excitatory neurons. Stuart and colleagues60 described an integrated data assays that included all embryonic datasets for downstream analysis. The clustering of cells was done by a shared nearest neighbour. The data was visualized with the use of the UMAP dimensional reduction.
The analysis of the embryonic datasets isjectory.
Trajectory inference and pseudotime calculations were done. The R library velocyto.R36 was used to estimate the RNA velocity. 10x output files were preprocessed using the command velocyto run10x. The velocyto.R-package was used for the estimation of RNA velocity.
The cells are mapped to clusters.
The five embryonic Seurat clusters from the integrated data assays that were located at the tip of the Monocle trajectory, as well as the Seurat clusters from the postnatal STICR dataset that were identified and annotated, were selected first. We used the Seurat FindVariableFeatures() function to find 1,855 genes that were variable features. We averaged the log-normalized expression in the clusters to create the model. The Pearson correlations between individual cells of the embryonic clusters were calculated. The significance of the assignment was determined by permuting the single-cell data for a random background and using empirical P values to assign each cell. We permuted the single-cell expression data 100 times. We kept track of the largest Pearson correlation to the model and calculated a P value for the cluster assignment by counting what fraction of correlation scores was larger than the one used for the assignment. In the final step, we mapped only cells with an FDR 1 TrackerSeq barcodes and retrieved their unique barcodes. The barcodes are highlighted in the picture. The arrows represent predictions of the future expression of genes. The mapping efficiency was used to pick the clusters.
The schematic shows different scenarios of how the brain could have cellular diversity. Different cell types could arise from specified or fate-restricted progenitor cells or from a common pool of progenitor cells. The fate of newborn cells can be determined by cell-intrinsic mechanisms or local cues at the progenitor level. The mechanisms that could drive the differentiation into different subtypes are called lineage-independent mechanisms. Independent mechanisms are not exclusive.
A report of collected data.
There are 20 differentially expressed genes for cell classes.
There are 20 differentially expressed genes for cell type clusters.
There are 20 differentially expressed genes.
You agree to abide by the Community Guidelines after submitting a comment. If you find something that does not comply with our guidelines, please flag it.
We use cookies to make sure that our website works properly, as well as some optional cookies to personalize content and advertising, provide social media features and analyse how people use our site. If you accept cookies, you give consent to the processing of your personal data, including transfer to third parties, in countries that do not offer the same data protection standards as the country where you live. Clicking on 'Manage settings' will show you more information about how your personal data is processed. Further information can be found in our privacy policy.
Page 2
A schematic and images of brain sections injected with STICR are included. The bars indicate time. Each dot is a clone. The one-way analysis of variance with Tukey post-hoc test was performed. A schematic of cell class origins is shown in the middle of the picture. There are light grey lines that link classes. The dotted line is a smooth local regression and the shadow is a 95% confidence interval.