Linguistics
Bayesian phylogenetics
We created a comparative dataset by combining dictionary search and fieldwork. It included 3,193 datapoints that represent 254 basic vocabulary concepts. This includes 98 Transeurasian languages. These concepts were derived from a combination of the Leipzig-Jakarta200 (ref. 43) and Jena200 (ref. 44) (ref. Based on recently published datasets45-46, the Turkic and Tungusic basic vocabulary are included. An inventory of basic vocabulary etymologies is available in Supplementary Data 2. This supports the coding of cognates.
Bayesian phylogenetic analyses were performed using binary data47 cognates. The data were collected in such a way that at least one cognate was found, it was determined that the data did not contain all zeros. This47 was corrected by ascertainment.
The following substitution models were considered, which govern the evolution of cognates along tree branches: the continuous time Markov chain, (CTMC), which assumes an equivocal rate of mutations; covarion which assumes both a slow and fast rate of mutations and model switching between them; and the pseudo Dollo Covarion model. This model is based upon the Dollo principle, that a cognate cannot appear more than once but can be lost multiple times. The literature contains detailed descriptions of the covarion and CTMC models47, as well as the pseudo Dollo model 48. We assume that every meaning class has its relative rate in order to capture variation between the rates of evolution for different words.
Although language is evolving at an average rate of one percent, there can be significant variation between branches in a tree's rates of change47,48. This variation can be captured by the uncorrelated relaxed clock49 as long as rates are log-normally distributed.
To describe the process of creating language, a birth death model is used. We allow ancestral nodes to be included in the tree because the data may contain ancient languages that are ancestral to current languages. The tree uses a fossilized birth death model50 as its prior. Age priors were used to inform language family node ages (Japonic 2100 +-175, Koreanic 800 +-175, Turkic 2100 +-175, Mongolic 775 bp+- 50, Tungusic 1900 +- 275). These calibrations can be supported by the chronological estimates proposed in linguistic literature (Supplementary Data 18, 18). These node age priors were found to help reduce the uncertainty in root age distribution.
The fit of various models was compared using nested sampling51 (Supplementary Data 18,). We concluded that the pseudo Dollo model with a relaxed clock and covarion with a relaxed clock had the best fit. Although both models produce comparable time estimates, covarion estimates have greater uncertainty, which means that they have higher 95% HPD intervals. The time estimates for the CTMC model with a relaxed clock are compatible, but they tend to be wider and have a higher average.
All posterior estimates were done using BEAST v.2.652 with adaptive coupled Markov Chain Monte Carlo (MCMC).53. The BEAST XML files (Supplementary data 19) provide detailed information about the models, priors and hyperpriors as well as the settings used to run them. Our Bayesian analysis results are presented as a dated tree of Transeurasian languages (Supplementary Data 24,).
Bayesian phylogeography
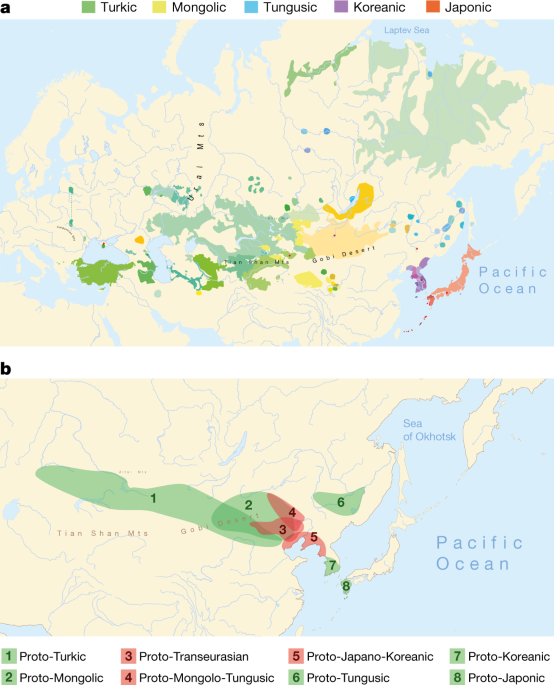
The dispersal of people through Eurasia could be described as a random walking, and diffusion on asphere54 is the best way to capture it. We performed a post-hoc analysis with the posterior tree from the lexical analysis to get an idea of the uncertainty involved in locating origins. While assigning point positions to the tips, we randomly selected trees from the posterior and estimated geographical parameters using MCMC. Even though this is a very restricted setup, it does not make it possible to identify the different hypotheses of geographical origin. Our analysis results are shown on a map (Supplementary Data 3). Bayesian phylogeography has limitations55,56. We complemented it with other homeland detection techniques such as linguistic paleontology and diversity hotspot principle in order to find a balanced location of the homelands for the root and nodes (Supplementary Data 4).
Linguistic Palaeontology
Comparative agropastoral vocabulary lists were created for each Transeurasian family: Turkic (Supplementary Data 5,a), Mongolic (5b), Tungusic (5c), Koreanic (6d), and Japonic (Supplementary Data 5,e). The linguistic reconstruction method is used to infer an ancestral state of a language that has not been attested based on data from a later time period. It can be applied to the corresponding words (Supplementary Database 5).
We used standard criteria that were based on the phonology of the word, its semantics and distribution to distinguish between borrowed and inherited correspondence sets. We identified distinct spatiotemporal patterns and cultural patterns in each category by dividing our dataset into inherited and borrowed subsistence vocabulary (Supplementary data 5).
Our subsistence vocabulary was subject to linguistic palaeontology. This is a historical comparative methodology that allows us to study human prehistory through correlating our linguistic reconstructions to archaeology information about the culture of ancient speech communities that used these terms. We were able to draw inferences about subsistence strategies for speakers of Transeurasian pro-languages during the Neolithic, Bronze Age (Supplementary Data 5), and we identified a plausible location as the homeland of the ancient speech community involved (Supplementary Data 4,).
Principle of diversity hotspot
We combined Bayesian and linguistic paleontology to estimate the location of ancient speech communities. This principle assumes that the homeland is the closest to the greatest amount of diversity in relation to the deepest subgroups within the language family. These areas were marked on the map, and used them to approximate the location of the first diversification of a proto-language (Supplementary data 4). This method has its limitations (Supplementary data 4), but it can provide a fairly reliable estimate of the location of an old speech community.
Archaeology
Database of archaeological data
We scored 172 cultural characteristics for 255 Neolithic–Bronze Age archaeological site or phases from the West Liao River basin (36), Amur (Jilin Heilongjiang, and inland Liaoning (32), Primorye ((4)), the Liaodong Peninsula (37), Eastern steppes (1) and Shandong peninsula (4). The Yellow River basin (2) was also included. The Korean peninsula (58), and the Japanese islands (85).
Sites that had multiple major cultural phases were not scored separately. These sites range in age from 8400 to 1700 bp. They include the Early Neolithic and Bronze Ages in northeast China, the Primorye's Middle Neolithic Zaisanovka and Bronze Age Mumun cultures, as well as the Late Neolithic and Bronze Age Final Jomon and Yayoi cultures of western Japan. Ceramics (70), stone tools (38) and buildings (9), as well as plant and animal remains (26) and shell and bone artifacts (17 and 12). Supplementary Data 6 (sheet 2) provides definitions of the scored features. Further discussion on scoring methods can be found at Supplementary Data 7. After reading published site reports and other literature, all features were scored as either present (1) or absent(0).
This database was used to analyze changes in the distributions of Neolithic artifacts and Bronze Age artifacts over time, in particular in relation to the spread Northeast Asian agricultural systems (Supplementary Data 7).
Bayesian phylogenetic analysis was used to analyse the cultural data in the archaeological database. There is a lot of phylogenetic work that uses archaeological data57. Some are distance-based58 and others parsimony-based58. Bayesian approaches have the advantage of being model-based and having solid mathematical foundations in probability theories. This allows us to estimate uncertainty around all estimates and allow us to integrate information from different sources (such as cognate and geographical data) in one analysis. BEAST is specifically designed to infer rooted time trees and uncertainty of time estimations. This sets it apart from other Bayesian programs that focus on unrooted trees. BEAST also supports models that are not currently available in other packages. This package is therefore recommended.
We used the same clock and substitution models for the lexical data to encode the cultural data. The pseudo Dollo model using a relaxed clock suits the data well (Supplementary Information 20). We also ran the analysis with the standard deviation set at 1. This was because the coefficient of variation for the relaxed clock was higher than 1, which indicated a lot of variation. It only slightly affected the time estimates.
Because of the uncertainty surrounding missing cultures and the large number of sampling dates, it was difficult to apply fossilized birth deaths prior. We instead chose the flexible Bayesian skyline plan60. Timing information is based upon archaeological finds' sampling dates. Because there is uncertainty about dating these finds, tip dates were uniformly collected in these intervals during MCMC. In line with previous archaeological studies61,62,63, we constrained the clades 'Xinglongwa-Zhabaogou-Hongshan' and 'Yabuli-Primorye' to be monophyletic (Supplementary Data 8). All analyses were done in BEAST v.2.652 with adaptive coupled MCMC53. The BEAST XML (Supplementary Data 21) contains details on priors, hyperpriors, and settings. The Bayesian analysis results are presented as a phylogenetic tree for Northeast Asian archaeological cultures (Supplementary Data 25, and interpreted by Supplementary Data 8.
Database of archaeobotanical information
We also compiled a database of archaeological features. (Supplementary Data 9). This list includes 269 samples: China, 82, Primorye 12; Korea 31; Japan (excluding Ryukyus), 120, Ryukyu Islands 24. The radiocarbon dates in this database were calibrated using OxCal v.4.4. To plot the distribution of cereals over time, we used kernel density mapping Supplementary Data 7. Supplementary Data 7 provided additional data for our databases, including published datasets on faunal remains64-65, dolmens66, and spindle howrls67.
Genetics
Labor procedures
The ancient DNA wet laboratory work (DNA extraction, library preparation) was done in a dedicated facility at the Max Planck Institute for the Science of Human History, MPI-SHH, and in an ancient DNA laboratory at Jilin University, following established protocols68. With 8-mer sequences at P5 and P7 Illumina adapters, a double-stranded library of DNA was created. To obtain adequate coverage, four individuals from China were shotgun-sequenced using the Illumina HiSeq X10 instrument. This was in the 150-bp paired end sequencing design. Three-thirds of the double-stranded libraries were created for 33 individuals in Japan and Korea. They were then characterized in the MPI–SHH by either shotgun sequencing or insolution capture at approximately 1.25 million nuclear single-nucleotide patterns (SNPs). Following initial screening, additional single-stranded library were created to retrieve more endogenous DNA. These libraries were shotgun-sequenced, in-solution-captured at approximately 1.2 million SNPs (Supplementary data 17), and sequenced on Illumina HiSeq4000 following manufacturer's instructions.
Sequence data processing
The EAGER v.1.92.55 program69 was used to process raw sequencing reads. AdapterRemoval v.2.2.070 was used to merge overlapping pairs and remove Illumina adapter sequences from the sequencing data. The merged reads were mapped with at least 30 bp to human reference genome (hs37d5 and GRCh37 with the decoy sequences) by BWA v.0.7.1271. DeDup v.0.12.260 was used to remove PCR duplicates. We used the trimbam function of bamUtils.1.0.1372 to mask 2 bp non-UDG libraries and 10 for half-UDG library reads on each end to minimize post-mortem DNA damage. SAMtools 1.360 used the mpileup function to pile up clean reads of both base quality (Phrede-scale quality) as well as mapping quality (Phrede-scale mapping quality). We called pseudo-diploid genotypes using the pileupCaller program (https://github.com/stschiff/sequenceTools ) against SNPs in the '1240k' panel73,74 under the random haploid calling mode. We used the masked BAM file for C/T and G/A; the unmasked BAM file was used for all other cases.
Reference datasets
Our ancient individuals were compared to three panels of genotype panels that are worldwide. One was based on Affymetrix HumanOrigins Genome-wide Human Origins 1 array (HumanOrigins; 593,124 autosomal sNPs)75, while the other panel is based on the '1240k’ panel73 and the Illumina' dataset76. These datasets were further enhanced by the addition of the Simons Genome Diversity Panel77. We also published ancient genomes (Supplementary Data 11).
An ancient DNA test
Multiple criteria were used to verify the authenticity of the newly published ancient genomes in Korea and Japan. We first used mapDamage version 2.0.678 to characterize the post-mortem chemical changes that are characteristic of ancient DNA. We used Schmutzi v.1.5.179 to estimate the mitochondrial contamination rates of all individuals. We also calculated the nuclear genome contamination rate for males using Schmutzi v.1.5.179. Because males only have one copy of the Xchromosome, any mismatches between bases that are aligned at the same polymorphic location beyond the level of sequencing error can be considered evidence of contamination. Fourth, we compared the positions of all damaged and reads in a Eurasia PCA to assess the possibility of West Eurasian contamination. We applied qpAdm74 to each individual to further characterize West Eurasian contamination using West Eurasian characteristic group such as Sintashta_MLBA and LBK_EN (see Supplementary data 17, 22 for more details).
Analysis of the population structure
A PCA was performed using the smartpca.v.1600082 with a set 2,077 current-day Eurasian people from the HumanOrigins dataset and the 1240kIllumina dataset with the option of 'lsqproject YES' or'shrinkmode YES. To measure the genetic affinity of two populations, since their divergence in Africa, we used outgroup-f3 statistics83.84 We used the 'f4mode : YES" function in admixtools31 to calculate f 4 stats. Both the f3 and f4 statistics were calculated with qp3Pop.
Genetic sex and uniparental haplogroup assignment
The molecular sex of the ancient samples was determined by comparing autosomes85 to X and Y coverages. Women would see an approximate equal ratio of X coverage to autosome coverage, and a 0. Men would get roughly half the coverage from X and Y as autosomes.
Mixture modeling with qpAdm
The qpWave/qpAdm framework was used to model the ancient individuals in the study (qpWave version.410 and QpAdm version.810) within the admixtools package74. The following 7 populations were used in the '1240k’ datasets as an outgroup ('OG): Mbuti and Onge, Iran_N. Villabruna. Karitiana. Funadomari Jomon. This set includes an African outgroup, Andamanese Islanders (Onge), early Neolithic Iranians (Iran_N), late Pleistocene European hunters-gatherers and Villabruna (Villabruna), native Karitiana from Brazil, a Tibetan Burman-speaking group from south China (Naxi), and ancient Japanese hunter-gatherers. (Supplementary data 13, 16).
Triangulation
Triangulation is a navigational technique that uses two measurements to determine a single point in space. It is used in qualitative research to describe a method that captures different dimensions of the same phenomenon using evidence from three scientific disciplines. Data collection, analysis, and the results of the research are done within the boundaries of each discipline to avoid any circularity. The final phase of triangulation is where the inferences drawn from the three disciplines are mapped onto each other using a variety of variables that describe the phenomenon. Triangulation has the purpose of increasing the credibility and validity by comparing the evidence from all three disciplines and identifying any correlations, inconsistencies or uncertainties across all perspectives.
We used triangulation to study the dispersal and distribution of Transeurasian languages. This method incorporated archaeology, genetics and linguistics to help us better understand the phenomenon. The methods described above were applied to different datasets to make independent inferences about a variety of variables, including location, chronology and migratory dynamics. We also considered continuity versus diffusion and subsistence (Supplementary Data 26, 26). Each discipline came up with the most plausible model that included these variables based on its own tools, which were either qualitative or quantitative and based on indirect evidence. A single discipline cannot resolve the question of farming/language dispersals. However, when taken together, the three disciplines enhance the credibility and validity. We were able to gain a better and more complete understanding of Transeurasian migration by combining the evidence from all three disciplines.
Summary of reporting
Additional information about research design can be found in the Nature Research Reporting Summary, which is linked to this article.