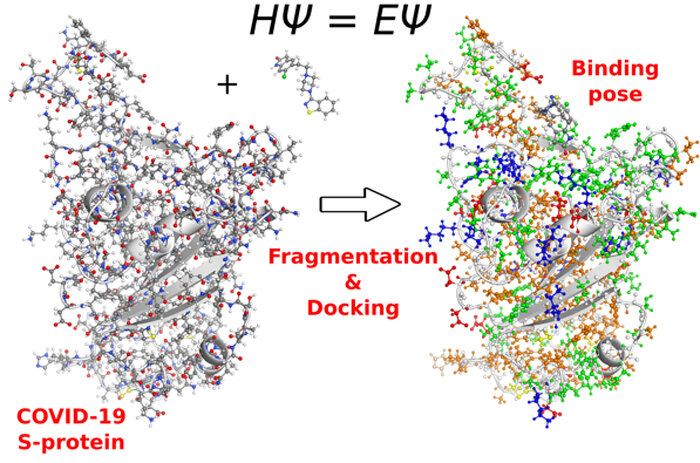
Oak Ridge National Laboratory researchers are using quantum refinement to pinpoint the binding sites for coronavirus spike protein. The refinement is part of a larger computational pipeline which scans millions of compounds to find the most promising ones for further testing. This illustration shows the process for protein structure preparation and fragmentation in order to refine quantum chemical docking. The COVID-19 S protein does not have a covalent bond because the ligand is not covalent. The colors on either side indicate fragments within the FMO formalism. Credit: Stephan Irle (ORNL).The disease has killed almost four million people since December 2019, when the first case was reported. Global infection has been reduced by a successful group of vaccines targeting the coronavirus's spike proteins. Supercomputers have accelerated the process of discovering new drug candidates that can be used to complement existing vaccines.Oak Ridge National Laboratory researchers are using the Stampede2 system at the Texas Advanced Computing Center to refine their screening for potential drug molecules that could disrupt spike protein binding to human cells.Refinement is only one part of a larger process that includes the search for entire databases with millions of compounds that have been approved by regulatory agencies, such as ZINC's public access database.In research published in December 2020 by the ACS Journal of Chemical Information and Modeling, the whole process was described as a supercomputer-driven pipeline to in silico drug discovery.The computational prescreening is done at the beginning and end of the pipeline process based on the structural model of target protein's individual atoms."We start by looking at the databases of molecules, and then we check the interactions between the molecules and the proteins. Stephan Irle (co-author of the drug pipeline study), is the leader of Oak Ridge National Laboratory's Computational Chemistry and Nanomaterial Sciences Group. These predictive quantum mechanical methods are used to predict the binding affinity of ligands and proteins.In a previous study at Chemrxiv, Jeremy Smith from ORNL performed 51,575 docking calculations involving six conformations of S-protein:ACE2 interface as well as 8,669 ligands from the ZINC15 databank. Based on their binding affinity to coronavirus spike proteins, the 3000 most promising candidates were selected.The Stampede2 supercomputer of the Texas Advanced Computing Center refines the results from a computational drug discovery pipeline to COVID-19. It uses a quantum chemical procedure that researchers at Oak Ridge National Laboratory developed. Credit to TACCThe top 3000 were then subjected to a gamut of quantum mechanics-based ranking refinement, binding analysis and a computationally challenging step that also included solvent effects.Irle and his colleagues validated the 100 strongest binding compounds and refined them using single-point energy calculations (FMO-MP2/PCM) by Irle. This resulted in a shortlist of 47 ligands that could be further tested. It also saved time as we concentrated on the most efficient inhibitors during experiment-theory feedback.TACC's Stampede2 supercomputer performed the computations. It took advantage of 48 Intel Skylake CPU cores (2 sockets) within one compute Node to perform a single geometry optimization. According to the June 2021 Top 500 rankings, the Stampede2 supercomputer is ranked #35 globally and #3 in U.S. academia."The Stampede2 performance is very high. The service and support were very responsive. "We had a very positive encounter," Van Quan Vuong, co-PI of quantum refinement, study coauthor on the drug discovery pipeline and a Ph.D. candidate supervised by Irle, The Bredesen Centre for Interdisciplinary Research and Graduate Education at the University of Tennessee, Knoxville.Irle said that for one ligand or protein complex, the binding energy (on Stampede2) can be obtained in just one to two hours. "If we have another computer node we can get the scoring function for a few hundred compound complicatedes in one day. This is a remarkable improvement on traditional DFT calculations.Jeremy Smith, ORNL, led the more recent work on the drug discovery pipeline. Molecular dynamics simulations were the mainstay of computation on the ORNL Summit IBM GPU based system. It is the second fastest supercomputer worldwide according to the June 2021 Top500 rankings. It was able to perform approximately 2.07 million physical docking calculations using a smaller database, and 2.4 billion docking calculations using the Enamine REAL compound database.Vuong and Irle are still developing the Stampede2 quantum mechanical refinement protocol. In the future, the researchers will investigate 15 spike protein clusters as well as refine the binding energies for 150 protein ligand compounds.Irle stated that "Reducing drug candidates and reliably narrowing to the most active species will eventually result in a quicker response to sudden emergent pandemics like the one we experienced with COVID-19." This is a novel approach because quantum mechanics has traditionally been considered too costly and an exotic area of computational drug discovery. We have now developed a method that allows us to complete quantum chemical analysis of the entire protein and ligand to determine reliable binding energies. This is a significant improvement over the traditional approach.Continue reading Early research using supercomputing on drug compounds could help combat the coronavirusMore information: A. Acharya et. al., Supercomputer-Based Ensemble Docking Drug Discovery Pipeline With Application to Covid-19. Journal of Chemical Information and Modeling (2020). Journal Information: Journal of Chemical Information and Modeling A. Acharya and colleagues, Supercomputer-Based Ensemble Docking Drug Discovery Pipeline With Application to Covid-19 (2020). DOI: 10.1021/acs.jcim.0c01010