Subscribe to Journal
Get full journal access for 1 year
$199.00
only $3.90 per issue
All prices are NET prices.
VAT will be added later in the checkout.
Rent or Buy article
Get time limited or full article access on ReadCube.
All prices are NET prices.
Additional access options:

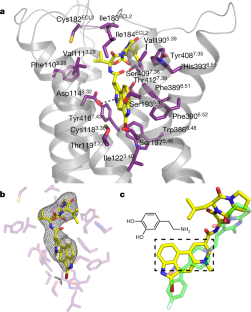

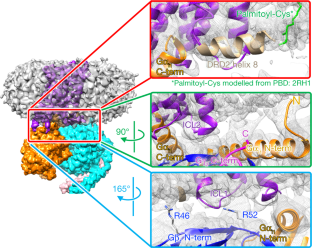
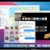
Structural data have been deposited in the PDB with coordinate accession number 6vms, and maps have been deposited in the Electron Microscopy Data Bank (EMDB) with accession numbers EMD-21243, EMD-21244 and EMD-21245. All other data generated or analysed during this study are included in this published article or are available from the corresponding authors on reasonable request. Madras, B. K. History of the discovery of the antipsychotic dopamine D2 receptor: a basis for the dopamine hypothesis of schizophrenia. J. Hist. Neurosci. 22, 62-78 (2013). PubMed Google Scholar Moritz, A. E., Free, R. B. & Sibley, D. R. Advances and challenges in the search for D 2 and D 3 dopamine receptor-selective compounds. Cell. Signal. 41, 75-81 (2018). Wang, S. et al. D 4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381-386 (2017). Koehl, A. et al. Structure of the µ-opioid receptor-G i protein complex. Nature 558, 547-552 (2018). Bayburt, T. H. & Sligar, S. G. Membrane protein assembly into nanodiscs. FEBS Lett. 584, 1721-1727 (2010). CAS Google Scholar Westfield, G. H. et al. Structural flexibility of the Gαs α-helical domain in the β 2-adrenoceptor Gs complex. Proc. Natl Acad. Sci. USA 108, 16086-16091 (2011). Rasmussen, S. G. F. et al. Crystal structure of the β 2 adrenergic receptor-Gs protein complex. Nature 477, 549-555 (2011). Wall, M. A. et al. The structure of the G protein heterotrimer G iα1β 1γ 2. Cell 83, 1047-1058 (1995). Simonds, W. F., Butrynski, J. E., Gautam, N., Unson, C. G. & Spiegel, A. M. G-protein βγ dimers. Membrane targeting requires subunit coexpression and intact γ C-A-A- X domain. J. Biol. Chem. 266, 5363-5366 (1991). Gallego, C., Gupta, S. K., Winitz, S., Eisfelder, B. J. & Johnson, G. L. Myristoylation of the Gα i2 polypeptide, a G protein α subunit, is required for its signaling and transformation functions. Proc. Natl Acad. Sci. USA 89, 9695-9699 (1992). Loisel, T. P. et al. Activation of the β 2-adrenergic receptor-Gα s complex leads to rapid depalmitoylation and inhibition of repalmitoylation of both the receptor and Gα s. J. Biol. Chem. 274, 31014-31019 (1999). Draper-Joyce, C. J. et al. Structure of the adenosine-bound human adenosine A 1 receptor-G i complex. Nature 558, 559-563 (2018). Cryo-EM data were collected at the University of Texas Southwestern Medical Center Cryo-EM Facility, which is funded by the CPRIT Core Facility Support Award RP170644. We thank E. Ross for providing plasmids for G-protein expression. This project was supported by the Edward Mallinckrodt, Jr. Foundation (Scholar Award to D.M.R.), the Welch Foundation (grant I-1770 to D.M.R. and grant I-1944-20180324 to X.-c.B.), the EPFL (to P.B.), the Swiss National Science Foundation (grant 31003A_182263 to P.B.), the Ludwig Institute for Cancer research (to P.B.), the Virginia Murchison Linthicum Scholar in Medical Research at University of Texas Southwestern (to X.-c.B.), CPRIT (RR160082 to X.-c.B.) and the US National Institutes of Health (R01-GM097207 to P.B. and R01-GM083118 to R.K.S.). J.Y. expressed and purified the DRD2-G i complex, collected cryo-EM data and carried out the cryo-EM reconstructions. K.-Y.M.C. carried out the computational design calculations on DRD2 mutants and performed in vitro experimental characterization of the designed DRD2 constructs. M.J.C. collected functional data on DRD2 constructs. M.H. carried out the molecular dynamics simulations of DRD2. P.K. assisted with DRD2 purification and testing of mutants. X.-c.B. supervised cryo-EM data collection and analysis. R.K.S. supervised the collection of functional data on DRD2 constructs and helped to write the manuscript. P.B. developed the strategy for computational design of a stabilized DRD2 active-state construct and supervised the computational design calculations. D.M.R. supervised the overall project, assisted with cryo-EM data analysis and wrote the manuscript. Correspondence to Roger K. Sunahara or Patrick Barth or Daniel M. Rosenbaum. P.B. is an inventor on a patent application (EP19189259.5) submitted by Ecole Polytechnique Fédérale de Lausanne on the design methods and designed protein variants herein. All other authors declare no competing interests. Peer review information Nature thanks R. Benjamin Free, Amy Moritz, David Sibley and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Publisher's note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. a, Energy landscape of three receptor variants with various energy differences in the resting (ligand-free) state ΔG resting, in the allosteric coupling controlling the inverse agonist efficacy ΔG inverse agonist and the stabilization of the receptor inactive state upon ligand stimulus. b, Active state occupancy as a function of ΔG resting and ΔG inverse agonist (the energy of inverse agonist binding), as determined by the Boltzmann law. c, Sensitivity of the receptors to inverse agonist binding. d- f, Rational design of DRD2 stabilized in the active state. DRD2 (D2) wild-type active state homology model ( d). The 120-residue-long ICL3 is represented as a dotted blue line. DRD2 designed with residue microswitches shown in spheres selectively stabilizing the active state structure ( e). DRD2 further stabilized in the active state through a de novo designed seven-residue stable ICL3 ( f). a, Competition binding of dopamine for [ 3H]spiperone on membranes containing hDRD2 WT (blue) or hDRD2 modified with nT4L and de novo-designed ICL3 (nT4L-hDRD2(ΔICL3); magenta). The log-transformed inhibition constant ( Ki) ± s.e.m. = −4.6 ± 0.04 for hDRD2 WT. log( Ki ± s.e.m.) = −4.6 ± 0.04 for nT4L-hDRD2(ΔICL3). n = 4 biologically independent experiments on Sf9 cell membranes. b, Agonist-stimulated [ 35S]GTPγS binding of nT4L-hDRD2(ΔICL3) (magenta) compared to hDRD2 WT (blue). Dose-responses using dopamine (closed magenta circles) or bromocriptine (BCT; open magenta circles) show log(EC 50 ± s.e.m.) = −7.5 ± 0.02 and −7.8 ± 0.06, respectively, for nT4L-hDRD2(ΔICL3) compared to log(EC 50 ± s.e.m.) = −7.0 ± 0.02 for dopamine on hDRD2 WT (open blue circles). n = 3 biologically independent experiments on HEK293T cell membranes. c, Saturation binding of [ 3H]spiperone to membranes containing hDRD2 WT with or without scFv16. Dissociation constant ( Kd) ± s.e.m. = 0.15 ± 0.03 nM for control. Kd ± s.e.m. = 0.14 ± 0.02 nM with scFv16. n = 4 biologically independent experiments on Sf9 cell membranes. d, Competition binding of dopamine for [ 3H]spiperone on membranes containing DRD2 WT with or without scFv16. log( Khigh ± s.e.m.) = −6.4 ± 0.3 and log( Klow ± s.e.m.) = −4.3 ± 0.08 for control. log( Khigh ± s.e.m.) = −6.5 ± 0.2 and log( Klow ± s.e.m.) = −4.3 ± 0.07 with scFv16. n = 4 biologically independent experiments on Sf 9 cell membranes. In all panels, each data point is displayed as the mean with the error bars showing ± s.e.m., with individual data points for each repeat represented in small symbols. a, G i stimulation by dopamine in Sf9 cells transformed with G i and hDRD2 baculoviruses. log(EC 50 ± s.e.m.) = −6.3 ± 0.1 for hDRD2 WT (blue). log(EC 50 ± s.e.m.) = −6.6 ± 0.3 for hDRD2 EM (orange). n = 4 biologically independent experiments on Sf9 cell membranes. b, Inhibition of dopamine-stimulated (10 μM) G i activation by the inverse agonist spiperone. log( IC 50 ± s.e.) = −8.6 ± 0.1 for hDRD2 WT (blue). log(IC 50 ± s.e.m.) = −6.3 ± 0.2 for hDRD2 EM (orange). n = 4 biologically independent experiments on Sf9 cell membranes. c, Modulation of basal G i activation by dopamine (open circles), BCT (closed circles) or spiperone (squares) with the hDRD2 EM (orange symbols) relative to hDRD2 WT (blue symbols). log(EC 50 ± s.e.m.) for dopamine (−6.6 ± 0.2), BCT (−7.6 ± 0.1) and spiperone (−7.6 ± 0.2) for hDRD2 EM. log(EC 50 ± s.e.m.) for dopamine (−7.0 ± 0.1) and BCT (−7.8 ± 0.2) for hDRD2 WT. n = 3 biologically independent experiments on Sf9 cell membranes. d, Apparent functional stability of agonist-bound hDRD2 constructs assessed by measuring the fraction of partially purified receptor-activating G i as a function of incubation time at 37 °C. Half-life ± s.e.m. = 27.6 ± 1.0 min for hDRD2 WT (blue). Half-life ± s.e.m. = 49.3 ± 1.1 min for hDRD2 with five designed mutations and designed truncated ICL3 (hDRD2(ΔICL3-5mut); orange). n = 3 biologically independent experiments. e, Apparent thermostability of agonist-bound hDRD2 constructs assessed by measuring the fraction of partially purified receptor-binding agonist as a function of temperature. Melting temperature ( Tm) ± s.e.m. = 30.9 ± 0.9 °C for hDRD2 WT (blue). Tm ± s.e.m. = 39.6 ± 0.3 °C for hDRD2(ΔICL3-5mut) (orange). n = 3 biologically independent experiments. In all panels, each data point is displayed as the mean with the error bars showing ± s.e.m., with individual data points for each repeat represented in small symbols. a, Superose 6 gel-filtration profile of hDRD2 EM-Gi-scFv16-HDL purified by M1 Flag affinity chromatography. b, Coomassie-stained PAGE of the isolated peak fraction from gel filtration. c, Overlaid Superose 200 gel-filtration profiles (in LMNG detergent) of different hDRD2 constructs purified by M1 Flag chromatography with saturating bromocriptine present. nT4L-hDRD2-5mut has the five thermostabilizing mutations of the EM construct, but has full-length ICL3. nT4L-hDRD2-ΔICL3 has the wild-type transmembrane region (no mutations) but the truncated ICL3. Receptors were purified as in Methods, but in the absence of co-transduced G-protein baculovirus. a, Representative 2D class averages. Scale bar, 100 Å. b, Gold-standard Fourier shell correlation (FSC) curves of the 3D reconstructions. The dashed lines intercept the y axis at a FSC value of 0.143. c, Image processing procedure with the final maps coloured based on local resolution. Ctf, contrast transfer function. a, Focused 3D classification subtracting T4L, the AH domain, scFv16 and rHDL/nanodisc density. b, Focused 3D classification subtracting all but the G protein. In a and b, the numbers below the images indicate the particle number in each 3D class. c, FSC curve showing the model-map correlation (focused map without T4L, the AH domain, scFv16 and rHDL/nanodisc density). The dashed line intercepts the y axis at a FSC value of 0.5. d, Representative hDRD2 transmembrane and ligand density. a, Solvent-accessible surface for the active conformation (purple) bound to bromocriptine (yellow sticks). b, Solvent-accessible surface for the inactive conformation (orange) bound to risperidone (green sticks), showing the deeper subpocket (PDB: 6cm4). a, Zoomed view of the TM6-TM7 interface on the cytoplasmic side of the receptor. Atomic interactions involving the designed Y6.40 were accurately predicted in the design model. The conformation of R3.50 is displaced in the experimental structure due to the binding of Gα i, which was absent in the model. The experimental structure is in purple and the predicted DRD2 conformation is in green. b, c, Conformational energy landscape of the wild-type ( b) and designed ( c) DRD2 constructed from multiple all-atom MD simulation trajectories of the receptor active state homology models (orange circle) in the absence of bound agonist. The conformational space is reported along two canonical active state structure metrics (TM3-TM6 distance and NPxxY rmsd to inactive state). The DRD2 wild-type relaxes primarily due to an inactive state conformation, whereas the designed variant remains in partially active state conformation displaying large TM3-TM6 distance. The antagonist-bound DRD2 crystal structure (PDB: 6CM4) is used as a reference for the inactive state (green square). a, AH domain density in the current structure. b, AH domain density in the rhodopsin-G i complex (PDB: 6cmo). c, Alignment of the Gα i subunit in the hDRD2 EM-G i (purple) and the rhodopsin-G i (blue) complexes. d, Alignment of the AH domain in the hDRD2 EM-G i (purple) and the rhodopsin-G i (blue) complexes. a, Surface potential of the hDRD2 (top) and G i (bottom) components shown separately. b, Surface potential of the complex, with the scale in eV. Calculation was done using the APBS plugin in PyMOL. Yin, J., Chen, K.M., Clark, M.J. et al. Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. Nature (2020). https://doi.org/10.1038/s41586-020-2379-5Data availability
References
t
Download references Acknowledgements
Author information
Affiliations
t
t
t
t
Contributions
Corresponding authors
Ethics declarations
Competing interests
Additional information
Extended data figures and tables
About this article

Cite this article
All Rights Reserved © 2024