The universally accessible application developed by the University of Minnesota Twin Cities will allow researchers to design better treatments for diseases like cancer.
A study was published in 2019. They made the application easy for non-experts to use and applied their findings to shed light on how the SARS-CoV-2 virus spreads.
The study can be found in Nature Communications and the app is free to use.
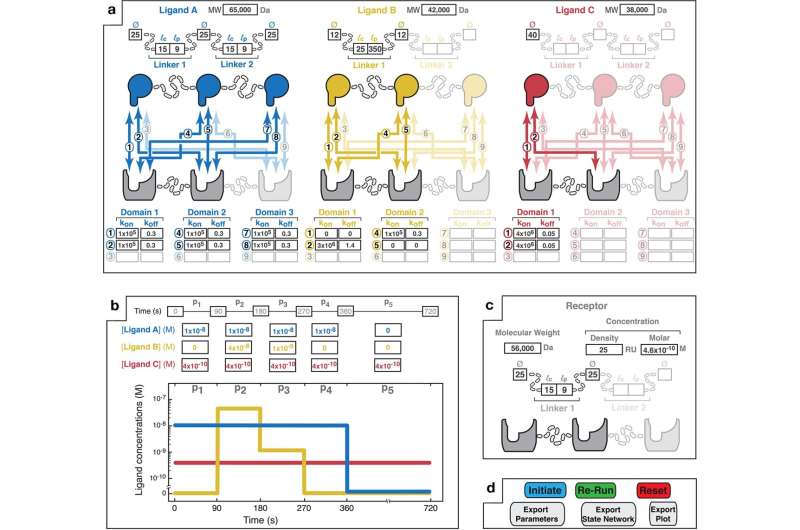
The strength, speed, and selectivity of multivalent interactions can be predicted with the help of the simulator.
According to the senior author of the paper, multivalent interactions are important in natural biological systems, and they are now starting to be creatively exploited for creating new therapeutic drugs that leverage their unique binding properties.
Multivalent drugs can be used to target cells in a way that isn't possible with standard, monovalent drugs, but there are many variables to consider in their design and much of the work in the field to date has been done through experimental trial and error. "MVsim allows us to make good predictions that can be used to design better therapies."
Many cancer drugs bind to cells that aren't meant to be targeted, which can cause unwanted side effects for the patient. It is possible to design drugs that target the cells in a tumors while not binding to other cells in the body.
The coronaviruses are another example. Scientists know that the virus is getting better at evading the immune system, but they don't know how it works. The University of Minnesota researchers were able to explore this process more in depth using theirMVsim technology.
The computational microscope that we have allows us to look under the hood and see what multivalent proteins are doing at amolecular level. It's difficult to capture this level of detail with a physical experiment. We can learn more about how these systems work, but we can also use this tool to design new multivalent interactions for diseases such as cancer.
The researchers plan to test some of the ways they have found to limit the spread of the disease.
More information: Bence Bruncsics et al, MVsim is a toolset for quantifying and designing multivalent interactions, Nature Communications (2022). DOI: 10.1038/s41467-022-32496-6The app is in the repository.
Journal information: Nature CommunicationsAll Rights Reserved © 2024